基于高通量数据进行HLA分型
一、简介
HLA即人类白细胞抗原(human leukocyte antigen),是编码人类的主要组织相容性复合体(MHC)的基因。其位于6号染色体的短臂上(6p21.31),包括一系列紧密连锁的基因座,与人类的免疫系统功能密切相关。其中部分基因编码细胞表面抗原,成为每个人的细胞不可混淆的“特征”,是免疫系统区分自身和异体物质的基础。
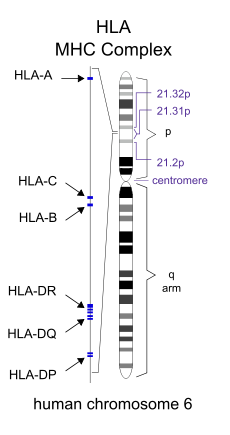
图1-1:HLA在染色体上的位置(来源于维基百科)
HLA基因可分为3大类:
- HLA-I型基因包括了HLA-A、HLA-B、HLA-C(HLA-E、F、G)等抗原基因;
- HLA-II型基因包含了HLA-DP、HLA-DQ、HLA-DR、HLA-DN和HLA-DO等系列基因;
- HLA-III型基因与前两型不同的是第三类MHC分子主要属于补体系统,参加不分别免疫。它可下分为C2、C4和BF等
HLA等位基因命名见下图所示:
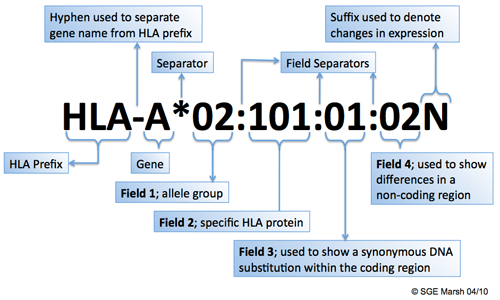
图1-2:HLA命名
- HLA是固定前缀;
- A代表基因名称,前缀和基因名称之间用短横线连接;
- *星号作为分隔符,分隔符之后是同一个基因的不同allel,由冒号分隔的多个部分构成,最多包含4个部分;
- 第一组02代表不同的血清学类型,根据血清学鉴定的结果对不同抗原进行分类;
- 第二组101代表一种特定类型的蛋白;
- 第三组的01代表发生在编码区的同义突变;
- 第四组的02代表发生在非编码区的基因突变;
- N为后缀,代表了蛋白的表达水平。
- 每一个HLA的Allel都会有一个唯一的ID,有时会在这些ID后面添加不同的后缀,不同后缀的含义不同(N、L、S、Q、C、A),具体解释如下:
- N表示该等位基因不表达;
- L代表low, 表示这种allel的表达水平相比正常水平要低;
- S表示蛋白产物不是位于细胞表面,而是一种可溶性的分泌蛋白;
- C表示蛋白产物不是位于细胞表面,而是位于细胞质中;
- A表示不确定这个allel的蛋白产物是否存在;
- Q表示这个allel会对其他等位基因的表达造成影响。
下面介绍两个基于高通量数据进行HLA分型的软件HALscan和OptiType。
二、HLAscan
HLAscan是一个基于比对的程序,在考虑read分布的情况下确定hla单倍型,可免费用于学术用途,商业下载和安装需要许可。github地址为:https://github.com/SyntekabioTools/HLAscan
下面介绍下它的安装使用:
wget -c https://github.com/SyntekabioTools/HLAscan/releases/download/v2.1.4/hla_scan_r_v2.1.4
chmod a+x hla_scan_r_v2.1.4
mv hla_scan_r_v2.1.4 ~/.local/bin/hla_scan # 为方便使用,将其移动到个人PATH路径下,并重命名
下载对应的测试集并解压:
wget -c https://github.com/SyntekabioTools/HLAscan/releases/download/v2.0.0/dataset.zip
unzip dataset.zip
需要注意的是,解压后不仅有测试集还有对应的数据库,下面我们使用其来进行HLA分型:
hla_scan -b data/NA12155.chr6.bam -d db/HLA-ALL.IMGT -v 38
其结果如下:
=====================================================
HLAscan v2.1
Report created
2021. 12. 16. 14:39:13
========================================================
HLA gene : HLA-A
# of considered types : 3182
----------- HLA-Types -----------
[Type 1] 01:11N EX3_3.29348_45 EX2_3.75926_100 EX4_24.0471_100 EX5_35.1966_100
[Type 2] 01:11N EX3_3.29348_45 EX2_3.75926_100 EX4_24.0471_100 EX5_35.1966_100
由于软件直接输出到终端上,所以平时我们运行的时候可以使用管道将输出重定向到文件中。除了bam文件外,HLAscan的输入还可以是fq文件:
hla_scan -l [fastq] (-r [fastq2]) -d [IMGT/HLA DB]
三、OptiType
OptiType 是一种基于整数线性规划的新型 HLA 基因分型算法,能够通过同时选择所有主要和次要 HLA I 类等位基因,从 NGS 数据中产生准确的 4 位 HLA 基因分型预测。github地址为:https://github.com/FRED-2/OptiType
其安装有几种方式,推荐使用conda进行安装,虽然也可以从源码编译,但是建议不熟悉编译的不要尝试,出错的话比较麻烦。
conda install optitype
或者
docker pull fred2/optitype
在使用OptiType进行HLA分析之前,建议先过滤出待分析序列中的HLA序列,这将能节省程序的运行时间,官方推荐使用RazerS3来进行比对,不过该软件会将序列读入到内存中,所以需要机器内存比较大。
在进行比对之前需要先下载对应的hla参考序列:https://github.com/FRED-2/OptiType/tree/master/data
测试数据:https://github.com/FRED-2/OptiType/tree/master/test
razers3 -i 95 -m 1 -dr 0 -o fished_1.bam hla_reference_dna.fasta sample_1.fastq # 进行比对
samtools bam2fq fished_1.bam > sample_1_fished.fastq # 将bam文件转为fq文件
rm fished_1.bam
对于双端文件的话,官方建议单独过滤双端文件,及fq1和fq2需要分别单独运行上诉命令。暂时不清楚这是为什么!
python OptiTypePipeline.py -i sample_1_fished.fastq (sample_2_fished.fastq) --dna -v -o output_dir -p out_prefix
上诉命令即可进行HLA分型,若是双端文件可以同时进行分析,我这里使用括号括起来,表示可选,如果你的是RNA数据,需要把--dna改为--rna
最终将会在结果目录下生成两个文件:*_coverage_plot.pdf 和 *_result.tsv。
结果如下所示:
A1 A2 B1 B2 C1 C2 Reads Objective
0 A*31:01 A*30:01 B*13:02 B*51:02 C*06:02 C*15:02 11426.0 10911.81
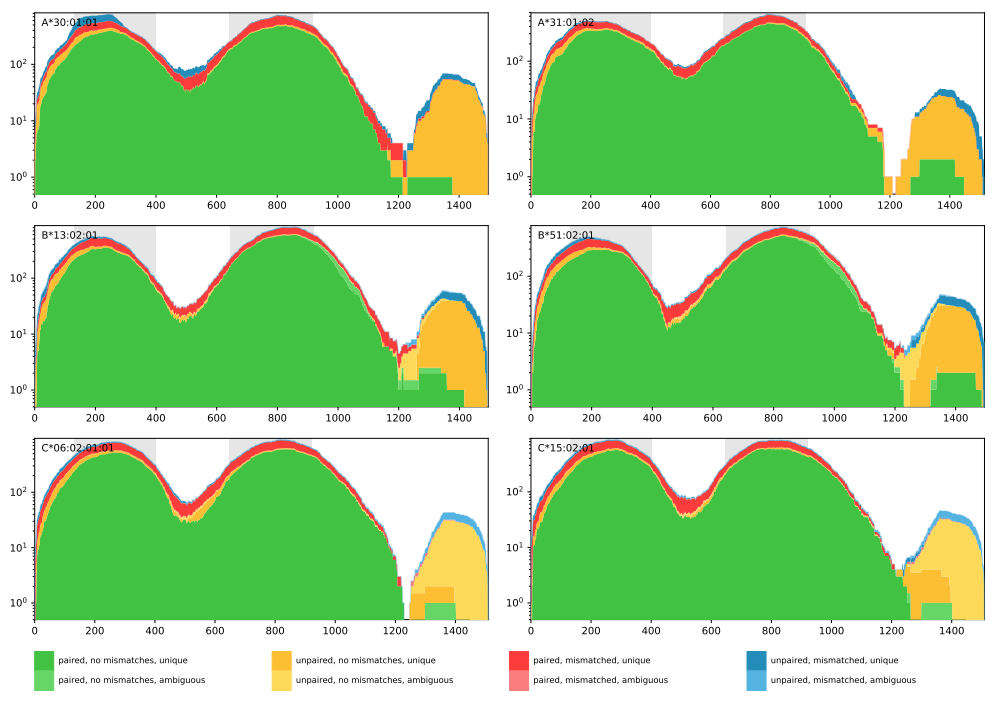
图3-1:*_coverage_plot 结果
四、参考




最新评论:
发表评论
电子邮件地址不会被公开。 必填项已用*标注